Serious incidents with medical device
Published:
Changes
Manufacturers, authorized representatives, importers, and distributors are legally obligated to report serious incidents involving their medical device.
Page contents
What is a Serious Incident
Serious incidents are any events that directly or indirectly have led, could have led, or can lead to any of the following:
-
Death of a patient, user, or another person
-
Temporary or permanent serious deterioration of a patient's, user's, or another person's state of health condition
-
Serious public health threat
Obligations of Market Participants
Manufacturers or their authorized representatives are obligated to report serious incidents according to the EU's post market surveillance system for medical devices. The report should be sent to the relevant authority in the country where the incident occurred. Serious incidents in Norway should be reported to the Norwegian Medical Products Agency (NoMA).
Incidents should be reported regardless of whether they are due to technical failures or deficiencies in the device, user manuals, labeling, use, or during maintenance of the device.
Manufacturers
It is the manufacturer's obligation to report serious incidents they have been notified of to relevant national authorities in the country where the incident occurred. The report must be submitted within specified deadlines. The manufacturer is required to conduct investigations to determine if their device may have contributed to the serious incident in any way. The manufacturer must also have a system in place to keep track of serious incidents reported with their device and monitor trends to detect if what is reported may be signs of systematic faults.
How to Report a Serious Incident
Authorized Representatives (AR)
Authorized representatives who have received complaints or reports from healthcare professionals, patients, or users about incidents believed to be related to devices they are designated for, must immediately forward this information to the manufacturer. They must also ensure that devices or device samples are made available for investigations.
Importers and Distributors
Importers and distributors who have received complaints or reports from healthcare professionals, patients, or users about incidents believed to be related to devices they have placed on the market, must immediately forward this information to the manufacturer and their authorized representative. Distributors can also report to importers.
Additionally, there are requirements for them to maintain records of received complaints, devices not in compliance with the requirements, as well as device recalls and withdrawals.
Incidents in Healthcare Facilities
Entities handling medical device have a national reporting obligation.
Healthcare facilities have an obligation to report serious incidents that have or may have a connection with the use of medical device.
Wish to report a serious incident? Use the link below.
Healthcare facilities report on melde.no
Private Individuals
If you as a private individual experience a serious incident, you are encouraged to contact the provider, distributor, or others from whom you obtained the relevant device. They are obligated to report further.
NoMA's work with serious incidents
The purpose of reporting is to provide NoMA with the opportunity to ensure that incidents are adequately investigated and to ensure that appropriate safety corrective actions are taken.
When NoMA receives reports of serious incidents with medical device from Norwegian healthcare facilities, they are promptly forwarded to the manufacturer. Regardless of where the manufacturer receives reports of serious incidents with their device, they are obligated to report to national authorities in the country where the incident occurred. NoMA receives reports when the incident occurred in Norway. The manufacturer investigates the incident and comes to a conclusion, which is documented in a final report sent to NoMA for review.
NoMA monitors serious incidents in collaboration with other European authorities.
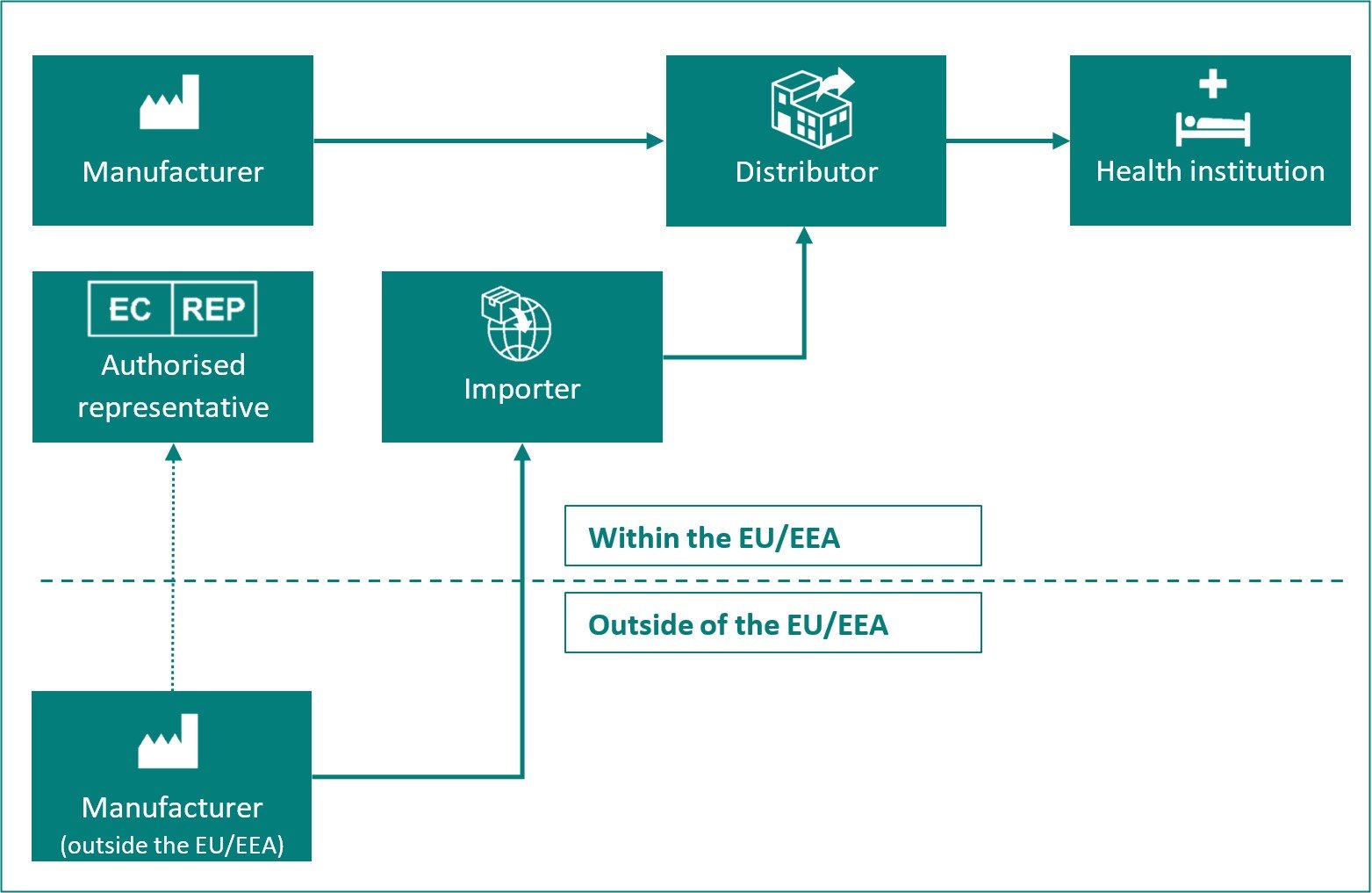
Overview of the different roles of economic operators:
| Economic operator |
Definition in MDR Article 2 and IVDR Article 2 |
| Manufacturer | A natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark. |
| Authorised representative | Any natural or legal person established within the Union* who has received and accepted a written mandate from a manufacturer, located outside the Union*, to act on the manufacturer's behalf in relation to specified tasks with regard to the latter's obligations under this Regulation. |
| Importer | Any natural or legal person established within the Union* that places a device from a third country on the Union* market. |
| Distributor | Any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service. |
| Health institution | An organisation the primary purpose of which is the care or treatment of patients or the promotion of public health. |
EU regulations on medical devices