Clinical trials applications with medicinal products containing GMOs
Published:
|
Updated:
Changes
- : Added figure
- : Added information regarding SNIF
- : Updated due to new regulation
Clinical trials involving medicinal products that contain genetically modified organisms (GMOs) must be assessed and approved under both the Clinical Trial Regulation and the Gene Technology Act before they can commence.
Page contents
Amendments to the Gene Technology Act that entered into force on 1 October 2025 have led to changes in how approval is sought for the use of GMO medicinal products in clinical trials.
All medicinal products that consist of or contain genetically modified organisms require approval if they are to be administered to patients who may leave the treatment facility.
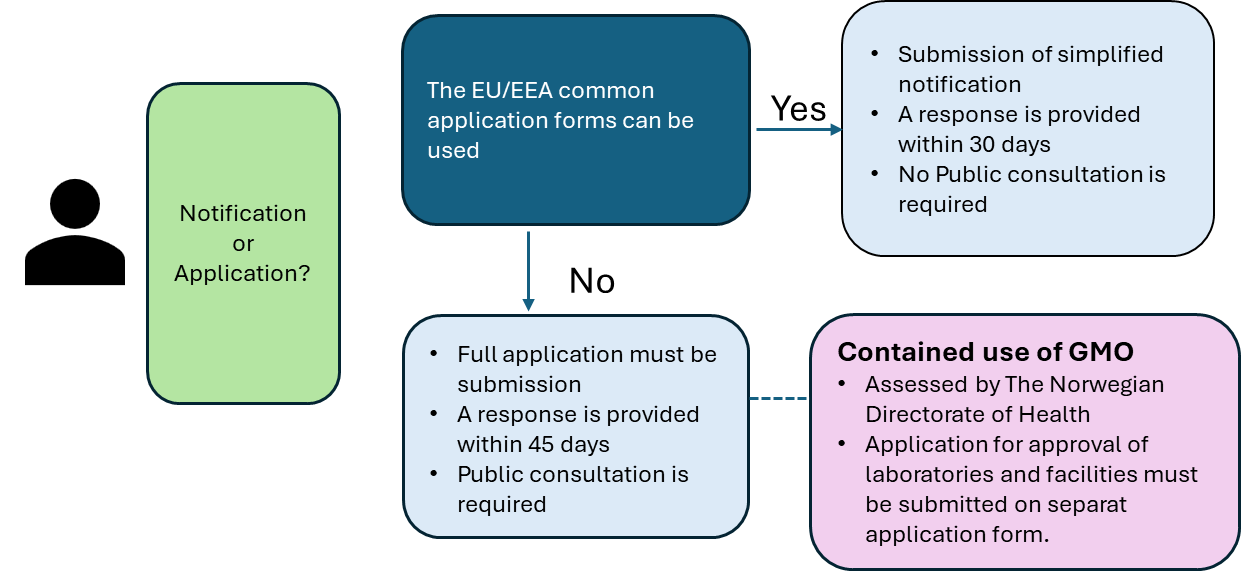
Depending on the type of GMO, the applicant must submit either a simplified notification or a full application to the Norwegian Medical Products Agency (NOMA). If a GMO medicinal product for human use is covered by the harmonised EU/EEA application forms (see below), it is sufficient to submit a notification.
If the medicinal product has a marketing authorisation, neither a notification nor an application needs to be submitted to NOMA. This also applies where the clinical trial concerns an indication other than the one for which the GMO medicinal product has been granted marketing authorisation.
All notifications and applications, including supporting documentation, shall be submitted to GMO@dmp.no. This is a joint mailbox for the Norwegian Environment Agency, the Norwegian Directorate of Health, and NOMA.
Please note that clinical trials involving medicinal products containing GMOs must be assessed and approved under both the Gene Technology Act and the Clinical Trial Regulation (CTR) before they can start.
Submission of simplified notification
Where the clinical trial concerns a GMO medicinal product that falls within one of the categories listed below, the common application forms may be used. The notification to NOMA must include:
- Completed application form
- Annexes as described in the form
- Confidential information and Summary Notification Information Format (SNIF)
- The SNIF must, in accordance with Council Decision 2002/813/EC, be completed and submitted to the European Commission register ’Food Chain (ESFC) platform’
If the Agency has questions or objections concerning the application, a response will be issued within 30 days.
AAV vectors
Guidance document: Good Practice on the assessment of GMO-related aspects in the context of clinical trials with AAV clinical vectors (pdf).
Human cells
Form: Common Application form for clinical research with human cells
genetically modified.
Guidance document: Good Practice on the assessment of GMO-related aspects in the context of clinical trials with human cells genetically modified.
Viral vectors
GMO applications where common application forms do not apply
The requirements below apply to GMO medicinal products for which the standard application forms referred to above cannot be used. The following shall be submitted to NOMA:
- Information as specified in Section 15 of the Regulation on Environmental Impact Assessment pursuant to the Gene Technology Act, Annex 1 Part A
- A health and environmental risk assessment, including conclusions, in accordance with the requirements set out in Annex 2 to the Impact Assessment Regulation.
- Documentation supporting the applicant’s environmental impact assessment of the GMO. This may include results from previous studies (raw data) and/or scientific literature.
- Summary Notification Information Format (SNIF), a summary of the application excluding confidential information. The SNIF may be submitted in Word or PDF format.
- The SNIF must, in accordance with Council Decision 2002/813/EC, be completed and submitted to the European Commission register ’Food Chain (ESFC) platform’
The decision under the Gene Technology Act will be issued before, or simultaneously with, the final assessment under the Clinical Trial Regulation. The initial assessment is normally provided within 45 days.
Contained use of GMOs
The Norwegian Directorate of Health will assess whether the rules on contained use of GMOs apply. The Directorate requires a description specifying whether the GMO:
- consists of genetically modified virus, bacterium, or cultured cell (specify type)
- is capable of replication or not
- is capable of transferring genetic material, etc.
Where the provisions on contained use of GMOs apply, the entity conducting the clinical trial must have approved laboratories and facilities for the contained use of GMOs.
Please also refer to the information available on Norwegian Directorate of Health.
Import and transport of GMOs
The Norwegian Environment Agency is the competent authority for decisions concerning the import and transport of GMOs pursuant to the Regulation on labelling, transport, import and export of genetically modified organisms. The Regulation specifies which GMOs require authorization under its provisions.
Where applications relate to clinical trials with medicinal products, the application may be submitted to gmo@dmp.no. NOMA will forward the application to the Norwegian Environment Agency. Further information is available on the Environment Agency’s website.