What are substitutable medicines?
Published:
Changes
Information about criteria for substitutability, substitutable medicines with different indications, substitution notes, substitution of H-prescription medicines, patent rights, and guidelines for the NOMA regarding medicine substitution.
Page contents
Medicines are substitutable when the following criteria are met:
- They contain the same active ingredient in equivalent amounts/same strength.
Only medicines with equivalent strength are suitable for automatic substitution in pharmacies. Substitution with a different strength, requiring the patient to split tablets or take more tablets than prescribed, should only occur exceptionally if the pharmacy does not have the correct strength in stock and the patient does not wish to wait for the medicine. - They have the same pharmaceutical form.
a. Different oral pharmaceutical forms with rapid release of the active ingredient are considered one and the same pharmaceutical form. For example, capsules and tablets with rapid release are considered equivalent. - They are bioequivalent or biosimilar.
- They are medically equivalent.
Bioequivalence
Two medicines are bioequivalent when their bioavailability is so similar that their effect and safety are essentially the same. Bioequivalence studies are conducted according to common European guidelines. These studies show whether the active ingredient is absorbed into the body to the same extent from both medicines.
In bioequivalence studies, all participants receive both the reference medicine and the generic medicine over several periods. Each participant is compared with themselves. Blood samples are taken from participants at regular intervals to measure the concentration of the medicine in the blood. Based on the analysis results, various pharmacokinetic values are calculated. The most common values are:
- AUC (Area Under the Curve): Total drug exposure over time
- Cmax (Maximum Concentration): The highest concentration after a dose
- Tmax (Time to Cmax): Time from dose intake to reaching Cmax
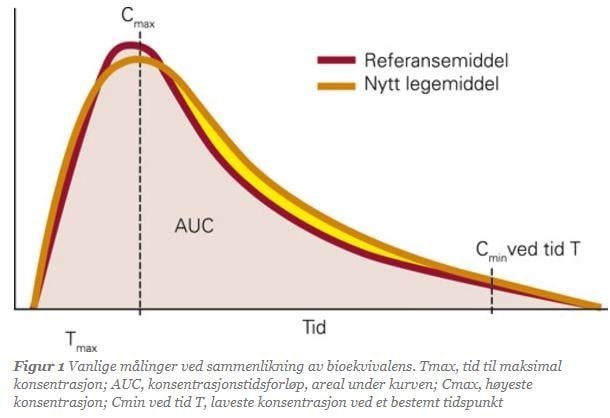
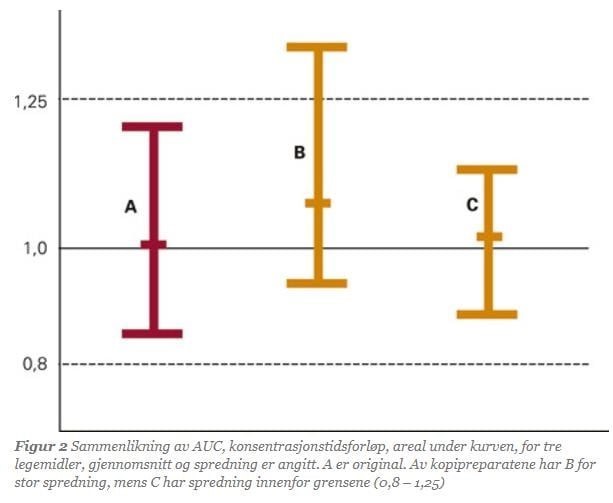
Two medicines are considered bioequivalent if the 90% confidence interval for the ratio of the mean values of AUC, Cmax, or Tmax for the reference and generic medicine lies within 0.8–1.25. When medicines meet this statistical requirement, they are bioequivalent. The studies are designed not only to demonstrate bioequivalence between medicines but also to show minimal variation in how different individuals absorb the medicine.
The same criteria for bioequivalence apply when the composition of a reference medicine is changed. The new composition is tested against the old one in a similar manner. Numerous studies have shown that generic medicines and their references have the same clinical effect. These studies show a very high correlation between the effects of generics and reference medicines when evaluated through bioequivalence studies.
Exceptions to the Requirement for Bioequivalence Studies
In some cases, medicines can be considered equivalent even if bioequivalence studies have not been conducted. For example, such studies are unnecessary for medicines administered directly into a vein, as there is always 100% absorption of the medicine into the body.
In other cases, documentation other than bioequivalence studies can be used to establish equivalence. Such documentation is carefully evaluated by the NOMAs expert group for equivalent substitution. Medicines without bioequivalence studies are sent for consultation before being potentially added to the substitution list.
Biosimilarity
A biosimilar medicine is a copy of a biological reference medicine. Biological medicines have a much more complex molecular structure than synthetic medicines because they are produced in living cells and not through chemical synthesis. It is not possible to create exact copies of biological medicines, which is why the term "biosimilar" is used for these, as opposed to "bioequivalent," which is used for generic medicines.
The rules for developing biosimilar medicines are similar to those for generic medicines and are based on the requirement of comparability: the active substance must be as similar as possible to the reference product. According to EU legislation, biosimilar medicines are granted marketing authorization because they are highly similar to the biological reference medicines. Recognized analytical and functional methods are used to test similarity with the reference medicine. These analyses are complemented by specially designed clinical studies, primarily pharmacokinetic ones. Biosimilar medicines can then be considered equivalent alternatives.
The European Medicines Agency (EMA) has confirmed that biosimilar medicines approved in the EU are interchangeable with the biological reference medicine and with other biosimilar medicines of the same reference medicine. See EMA's assessment of biosimilarity.
Medical equivalence
In addition to assessing bioequivalence/biosimilarity, the NOMA also evaluates whether the medicines are medically equivalent. This means assessing whether it is safe to include the medicines on the substitution list, taking into account other factors, such as:
- Disease/patient group
- Risk of serious problems with improper use
- Need for special equipment, such as injection pens or inhalation devices
- Small differences in drug absorption between patients that could cause problems
Only medicines that are both bioequivalent/biosimilar and medically equivalent are included on the substitution list.
Substitutable medicines with different indications
It is important to note that substitutable medicines are medically equivalent even if there are differences in the package leaflet and product description, such as different indications and age recommendations. When NOMA assesses whether medicines should be substitutable, we specifically evaluate whether such differences hinder safe substitution.
Substitution annotations
Substitution notes establish any limitations on the substitutability of medicines.
One type of substitution note is "limited substitution." Medicines with this note can only be substituted at the start of treatment. When the patient later collects the same medicine, it should not be substituted unless the physician has approved the substitution.
Certain medicines have limited substitution because they have a narrow therapeutic window or other issues related to dosage and effect. For example, epilepsy medicines have limited substitution. For individuals with epilepsy, even small differences in the blood concentration of the medicine can have serious consequences, such as seizures or side effects. It is particularly important that epilepsy medicines provide a consistent and predictable effect, as the consequences of misuse can be significant. There are no substitution restrictions when the medicine is used for other conditions.
Other substitution notes are related to indication patents or other substitution-related considerations that the pharmacy should be aware of when dispensing the medicine. Substitution notes are displayed to pharmacy staff when the medicine is dispensed.
Substitution of Hospital-Financed Medicines (H-Prescription Medicines)
The substitution scheme is intended to promote price competition and limit the growth of public expenditure on medicines. Medicines used exclusively in hospitals are also included in the substitution list. Although H-prescriptions are not funded by public health insurance, they are included in the substitution scheme as this is a service that benefits hospitals. Substitution of H-prescription medicines is strengthened by the medicine authority's assessment that the relevant medicines are substitutable.
Medicine substitution of hospital-financed medicines (H-prescription medicines) is covered by a settlement agreement between the regional health authorities and the Pharmacy Association.
Patent Rights
Patent disputes may arise concerning a single indication or the medicine as a whole. Such disputes must be resolved by the private parties involved and are outside NOMAs assessment of whether the medicines can be included on the substitution list. NOMA will not delay listing a medicine on the substitution list until the dispute is resolved.
Guidelines for NOMAs work on substitutable medicines
The NOMA assesses the equivalence and substitutability of all medicines that are granted marketing authorization and pricing. Medicines that, according to the guidelines, should be assessed by the expert group are reviewed in meetings held once a month. The expert group includes members with expertise in pharmacy, medicine, regulatory affairs, and law.
Certain cases must be sent for consultation before the expert group can make a formal decision on inclusion in the substitution list.
Guidelines for inclusion on the substitution list
Pharmacies can substitute prescribed medicines with generically equivalent medicines, biosimilar equivalent medicines, or parallel-imported medicines if the Ministry of Health and Care Services (via NOMA) has approved the medicines as substitutable, in accordance with the Pharmacy Act § 6-6, second paragraph.
The NOMA assesses all medicines with the same active ingredient for inclusion on the substitution list, regardless of the basis for marketing authorization. Indications with patent protection are excluded from the substitution scheme in pharmacies.
Medical equivalence is assessed based on documentation, which may include pharmacokinetic equivalence, bioequivalence, disease/patient group, risk of serious problems with improper use, need for special administration equipment, training needs, or whether small differences in absorption in individual patients could cause medically significant problems.
Definitions
- Bioequivalence Studies: Studies conducted to demonstrate that medicines are bioequivalent.
- Bioequivalent Medicines: Medicines containing the same active ingredient and absorbed into the body in such a similar way that they have the same therapeutic effect.
- Bioavailability: The total absorption of a medicine in the body.
- Biowaiver: Permission to omit bioequivalence studies under certain conditions.
- Substitutable medicines: Medically equivalent medicines that can be substituted at the pharmacy.
- Substitution mist: The official Norwegian list (regulation) of medicines considered medically equivalent and substitutable at pharmacies.
- Pharmacodynamics: The effects of a medicine in the body.
- Pharmacokinetics: The absorption, distribution, metabolism, and excretion of a medicine in the body.
- Generic medicine: A medicine with the same qualitative and quantitative composition of active ingredients and the same pharmaceutical form as the reference medicine, with bioequivalence demonstrated through relevant bioavailability studies.
- Biosimilar medicine: A copy of a biological reference product (original).
- Excipients: Substances in a finished medicine in addition to the active ingredient, such as binders, fillers, colorants, or pH regulators.
- Indication patent: A patent on the use of an active ingredient for a specific indication.
- NOMAs expert group for equivalent substitution: A multidisciplinary group covering areas such as medicine and pharmacy, evaluating the medical equivalence of medicines and advising NOMA leadership on inclusion in the substitution list.
- Marketing authorization (MA): The right to sell and market an approved medicine in Norway.
- Medically equivalent medicines: Medicines that NOMA considers safe to list on the substitution list, taking all factors into account.
- Substitution note: Notes attached to certain medicines on the substitution list, specifying any limitations on their substitutability.
- Parallel-Imported medicine: A medicine with valid MA in an EU/EEA country, imported via a third country by an independent importer and marketed in competition with a directly imported equivalent.
- Original medicine: The original medicine that has been patent- and/or competition-protected for a period.
- Well-established use: A medicine that has been extensively used in patients for at least ten years and is approved based on the experience accumulated.
- Active ingredient: The chemical or biological compound that provides the therapeutic effect in a finished medicine.
Requirements for marketing authorization
Only medicines that have marketing authorization (MA) in Norway can be included on the substitution list.
Evaluation of medicines for inclusion on the substitution list
If there are multiple equivalent medicines with the same active ingredient, they must be evaluated for inclusion on the substitution list. Differences in salts, esters, isomers, and other variants of the active ingredient, as well as differences in excipients (e.g., the amount and type of solvent), are only considered if they affect the safety or efficacy of the medicines. Variations in approved medical indications, differences in administration equipment, and other differences between the medicines do not prevent inclusion on the substitution list but may require special consideration.
This applies regardless of the basis for marketing authorization.
Substitution annotations
The substitutability of certain medicines may be limited in various ways. NOMA then issues a "substitution note" linked to the medicine in the substitution list. Medicines that can only be substituted at the start of treatment receive such a note. Substitution notes can also be linked to specific indications due to regulatory or medical considerations. Additionally, substitution notes may include information about special considerations pharmacies must be aware of when dispensing the medicine.
Patent
Patent disputes, whether they concern a single indication or the medicine as a whole, must be resolved by the private parties involved and are outside NOMAs assessment of whether medicines can be included on the substitution list. NOMA will not delay listing the medicine on the substitution list until the dispute is resolved.
Hearings on inclusion of medicines in the substitution list
The substitution list is a regulation, and NOMA must send the inclusion of medicines for consultation. However, consultation may be waived if it is deemed "clearly unnecessary."
A consultation period of three weeks is typically set.
Hearings may be waived for inclusion on the substitution list:
- For generic medicines where bioequivalence with the reference medicine has been demonstrated.
- If bioequivalence studies have not been conducted in two cases:
- When the regulations allow exemptions from the requirement for bioequivalence studies as a basis for MA.
- When an MA holder has been granted an exemption from the requirement for bioequivalence studies (biowaiver) during the processing of the MA application.
Medicines that can be included on the substitution list without special review by NOMAs expert group for equivalent substitution:
- Parallel-imported medicines and duplicate medicines.
Inclusion may be reviewed by the expert group if specific issues need to be addressed. - Medicines approved under the Medicinal Products Regulation § 3-9 (Article 10 in Directive 2001/83), except for:
- Medicines approved under § 3-12 (Article 10a) (well-established use), § 3-9 (4) (Article 10.3) (hybrid application), and biosimilar applications (Article 10.4).
- Medicines in the following ATC groups:
- A10 (Diabetes medicines)
- B01 (Antithrombotic agents)
- J06 (Immune sera and immunoglobulins)
- J07 (Vaccines)
- L01 (Antineoplastic agents)
- L03 (Immunostimulants)
- L04 (Immunosuppressants)
- N03 (Antiepileptic agents).
- Medicines with primarily local effects (e.g., creams, eye drops, nasal sprays, etc.).
- Medicines with special administration equipment (e.g., inhalers, injection pens, infusion bags, etc.).
- Medicines approved with a European reference medicine.
- Medicines with indication patents.
- Medicines that must be sent for consultation (see 2.4).
- Medicines and medicine groups where NOMA has established practices through previous evaluations in similar cases.