Søknad om klinisk utprøving av GMO i legemidler til mennesker
Publisert:
|
Oppdatert:
Endringer
- : Lagt til informasjon om opprettelse av SNIF i E-Submission Food Chain platform (ESFC).
- : Lagt til figur, mindre endringer i teksten
- : Mindre språklige justeringer
- : Lagt til setning om at endringen trådte i kraft 01. oktober 2025
- : Teksten er oppdatert som følger av revidert genteknologilov
Kliniske utprøvinger med legemidler som inneholder GMO må være vurdert og godkjent etter både regelverket for kliniske studier og genteknologiloven før de kan starte.
Innhold på siden
Endringer i genteknologiloven som trådte i kraft 1. oktober 2025 har ført til endringer i hvordan man søker godkjennelse for bruk av GMO-legemidler i kliniske studier.
Alle legemidler som består av eller inneholder genmodifiserte organismer krever godkjenning dersom de skal gis til pasienter som kan forlate behandlingsfasiliteten.
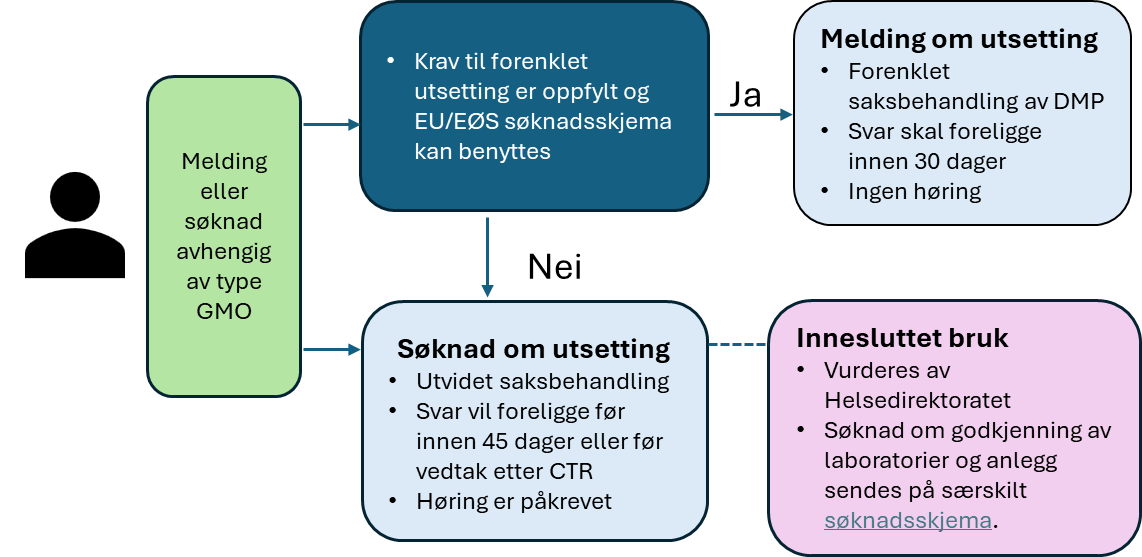
Avhengig av type GMO, skal søker sende inn en forenklet melding eller en søknad til Direktoratet for medisinske produkter (DMP). Dersom et GMO-legemiddel til mennesker omfattes av EU/EØS sine ferdige søknadsskjemaer (se nedenfor), er det tilstrekkelig å sende inn en melding.
Dersom legemidlet har markedsføringstillatelse skal det ikke sendes inn hverken melding eller søknad til Direktoratet for medisinske produkter. Dette gjelder også når den kliniske utprøvingen skal gjøres på en annen indikasjon enn det GMO-legemiddelet har markedsføringstillatelse for.
Alle meldinger og søknader med tilhørende dokumentasjon skal sendes til GMO@dmp.no.
Husk at kliniske utprøvinger med legemidler som inneholder GMO må være vurdert og godkjent etter både genteknologiloven og regelverket for kliniske studier (CTIS) før de kan starte.
Forenklet melding om bruk av GMO-legemidler
Dersom studien gjelder et GMO-legemiddel som faller inn under en av kategoriene nedenfor kan søknadsskjemaene brukes. Meldingen til DMP skal inneholde:
- Utfylt skjema
- Vedlegg som beskrevet i skjemaet
- Konfidensiell informasjon og kopi av Summary Notification Information Format (SNIF), en kortfattet oppsummering av søknaden uten konfidensiell informasjon. SNIF kan sendes inn enten i word- eller PDF-format.
- SNIF opprettes og sendes inn elektronisk til Europakommisjonens register for GMO-søknader E-Submission Food Chain platform (ESFC).
Dersom Direktoratet for medisinske produkter har spørsmål eller innvendinger til søker, vil svar sendes ut innen 30 dager.
AAV Vektorer
- Skjema: Common application form for investigational medicinal products for
human use that contain or consist of AAV vectors (pdf). - Tilhørende veiledningsdokument: Good Practice on the assessment of GMO related aspects in the context of clinical trials with AAV clinical vectors (pdf).
Humane celler
- Skjema: Common Application form for clinical research with human cells
genetically modified (pdf). - Tilhørende veiledningsdokument: Good Practice on the assessment of GMO-related aspects in the context of clinical trials with human cells genetically modified.
Virusvektorer
GMO-søknad der ferdige søknadsskjema ikke gjelder
Kravet under gjelder for GMO-legemidler der man ikke kan bruke ovennevnte skjemaer. Vi ber om at følgende sendes inn til DMP:
- Informasjon som gitt i §15 i Forskrift om konsekvensutredning etter genteknologiloven (Ekstern lenke) (heretter omtalt som KU-forskriften), vedlegg 1 del A.
- Helse- og miljørisikovurdering med konklusjoner som følger kravene i vedlegg 2 i KU-forskriften.
- Dokumentasjon som underbygger søkers konsekvensutredning om GMO-ens egenskaper. Dette kan bl.a. være resultater fra tidligere forsøk (rådata) og/eller vitenskapelig litteratur.
- Kopi av Summary Notification Information Format (SNIF) i pdf- eller word-format. SNIF vil legges ut på høringssiden og vil være offentlig tilgjengelig.
- SNIF opprettes og sendes inn elektronisk til Europakommisjonens register for GMO-søknader E-Submission Food Chain platform (ESFC).
Svar på søknaden etter genteknologiloven vil bli sendt før, eller samtidig med endelig vurderingen av søknaden etter legemiddelregelverket. Første vurdering foreligger normalt innen 45 dager.
Innesluttet bruk av GMO
Ved mottak av en GMO-søknad vil Helsedirektoratet vurderer om reglene for innesluttet bruk av GMO kommer til anvendelse. Helsedirektoratet krever en beskrivelse som angir om GMO-en:
- er en genmodifiserte virus, bakterier eller celler i kultur (hva slags)
- kan replikere eller ikke
- kan overføre genetisk materiale etc.
Hvis reglene om innesluttet bruk av GMO kommer til anvendelse, må virksomheten som skal gjennomføre utprøvingen ha godkjente laboratorier og anlegg for innesluttet bruk av GMO. Se mer informasjon på Helsedirektoratets hjemmeside (ekstern lenke).
Import og transport av GMO
Miljødirektoratet er vedtaksmyndighet for import og transport av GMO etter forskrift om merking, transport, import og eksport av genmodifiserte organismer (Ekstern lenke). I forskriften finner du oversikt over hvilke GMO som krever godkjenning etter denne forskriften. Når søknader gjelder legemiddelutprøvinger kan søknaden sendes til gmo@dmp.no. DMP vil da videreformidle søknaden til Miljødirektoratet. Se mer informasjon på Miljødirektoratets side (Ekstern lenke).