Søknad om klinisk utprøving av legemidler til mennesker
Publisert:
|
Oppdatert:
Endringer
- : Faglig gjennomgang. Mindre språklige endringer.
- : Oppdatert brutt lenke på nettsiden
- : Teksten er gjennomgått og forbedret for bedre å gjenspeiler regelverk og praksis.
- : Oppdatert informasjon om gebyrer
Informasjon om saksbehandling, språkkrav, regler for offentliggjøring og konfidensialitet.
Innhold på siden
Før søknaden kan opprettes
Clinical Trials Information System (CTIS) er en felles nettportal for innsending av søknader relatert til kliniske utprøving av legemidler til mennesker i EU og EØS.
For informasjon om tilgang til systemet, nettbasert opplæring og generell informasjon om saksbehandling i CTIS, gå til vår nettside Clinical Trials Information System (CTIS).
Veiledning og råd til bedrifter og akademia
DMP bistår bedrifter og akademia med veiledning og råd innenfor utvikling av medisinske produkter, klinisk utprøving og helseøkonomi. DMP veileder også apotek, grossister og tilvirkere på legemiddelområdet.
Les mer
Søknad om ny klinisk utprøving
En søknad om klinisk utprøving består av to deler. Del I består av dokumentasjonen som i en multinasjonal studie er felles for alle deltakende medlemsland, blant annet protokoll, Investigators Brochure (IB) og produktinformasjon. Del II inneholder dokumentasjon som er tilpasset hvert enkelt medlemsland, blant annet pasientinformasjon, samtykkeskjema og forsikringsbevis.
Dersom den kliniske utprøvingen skal gjennomføres i flere medlemsland, må sponsor angi i søknaden, hvilket av disse landene som skal ha rollen som Reporting Member State (RMS). Det er RMS som leder og koordinerer valideringen og utredningen av dokumentasjon i del I av søknaden.
En søknad om klinisk utprøving må alltid inneholde del I. Del II kan sendes inn for ett eller flere medlemsland samtidig med del I. Alternativt kan sponsor sende inn del II til de ulike medlemslandene i etterkant, men senest to år etter at del I er godkjent. Dersom sponsor ikke sender inn del II innen to år etter konklusjonen for del I, bortfaller søknaden. Del I vurderes av både DMP og REK KULMU, mens del II vurderes kun av REK KULMU.
Mer informasjon om:
Saksbehandling av søknad om klinisk utprøving
Forordning (EU) Nr. 536/2014 bestemmer tidslinjene for saksbehandlingsprosedyrene i CTIS. Tidslinjene kan variere avhengig av hvilken type søknad som skal behandles (initiell, vesentlig endring, legge til medlemsland etc.). For informasjon om de ulike tidslinjene, se EMAs veiledningsdokument: CTIS Sponsor Handbook (PDF).
Etter at en søknad er sendt inn kan man få tilgang til prosedyrens estimerte tidslinje under «timetable» i søknadsdossieren i CTIS.
Fristene i CTIS er basert på kalenderdager. Merk at CTIS kun tar hensyn til nasjonale helligdager til medlemsstaten som har rollen som RMS. Fristene er dynamiske, det vil si at etterfølgende frister endres dersom sponsor eller myndigheter leverer tidligere enn fristen som er angitt i CTIS. Det sendes ikke varsel på epost når myndighetene sender ut merknader eller vedtak i CTIS, så det er viktig å sjekke systemet regelmessig.
Det er ikke teknisk mulig å forlenge fristene i CTIS. Dersom frister ikke overholdes, vil søknaden utgå («lapse»). Det er ikke mulig å gjenåpne en søknad som har utgått, så denne må innsendes på nytt.
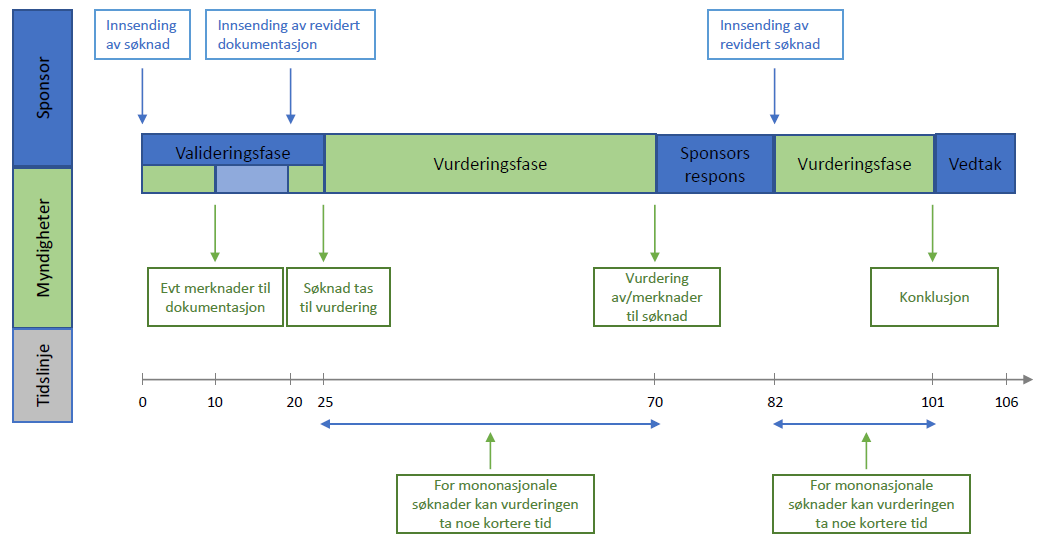
Saksbehandling av en søknad om klinisk utprøving er inndelt i 3 faser:
Valideringsfasen
-
Myndighetene har 10 dager på å gjennomgå søknadsdokumentasjonen. Vi sjekker at dokumentasjonen er fullstendig og korrekt lastet opp.
-
Ved spørsmål til søknaden vil sponsor vanligvis få 10 dager til å svare.
-
Sponsor vil motta en beslutning på om søknaden er valid innen 25 dager etter innsendelse.
Vurderingsfasen
-
Utredningen av en søknad om klinisk utprøving tar fra 38 til 45 dager (avhenger av om studien er mononasjonal eller multinasjonal).
-
Dersom myndighetene har spørsmål til søknaden (Request for information, RFI), vil dette forlenge vurderingsfasen med maksimalt 31 dager - 12 dager for sponsor til å svare på spørsmål og ytterligere 12-19 dager for vurdering av sponsors svar.
-
Dersom søknaden involverer utprøving av et legemiddel som er definert som avansert terapi (ATMP) kan RMS forlenge vurderingsfasen med maksimalt 50 dager.
-
Sponsor vil motta konklusjon på del I av søknaden i CTIS.
-
Utredning av del II vil gå parallelt med del I.
Beslutningsfasen (Decision)
-
Sponsor vil motta et endelig nasjonalt vedtak innen 5 dager etter at både Del I og Del II har blitt konkludert. Vedtaket utstedes i CTIS.
-
En søknad om klinisk utprøving kan bli godkjent, godkjent med vilkår eller avslått.
Regler for publisering i CTIS
Informasjon om kliniske studier som er søkt i CTIS gjøres tilgjengelig i den offentlige CTIS-portalen i tråd med CTIS Transparency Rules (pdf).
I hovedsak gjelder følgende for publisering:
-
Strukturerte data (med enkelte unntak) vil publiseres i sin helhet.
-
Publisering av søknadsdokumenter er begrenset til enkelte “nøkkeldokumenter”, i hovedsak protokoll, protokollsynopse, pasientinformasjon, samtykkeerklæring og studierapport.
-
Nøkkeldokumentene må sladdes og sendes inn i to versjoner (for publication og not for publication) dersom de inneholder personlig og/eller sensitiv informasjon.
Det er sponsors ansvar å sikre samsvar med forordning (EU) nr. 2016/679 og forordning (EU) nr. 2018/1725 ved opplasting av dokumenter og behandling av personopplysninger i CTIS.
Tidspunktet for offentliggjøring av dokumentasjon styres av hvilken kategori den kliniske utprøvingen er i:
-
Category 1 trials - Pharmaceutical development
-
Category 2 trials- Therapeutic exploratory and confirmatory clinical trials
-
Category 3 trials - Therapeutic use clinical trials
Det er sponsor som definerer hvilken kategori studien tilhører ved innsendelse av søknad.
Gebyrer
Fakturainformasjon må lastes opp under “Proof of Payment of fee" under “Form", i del I av søknaden i CTIS. Det vil bli sendt en faktura etter at søknaden er validert.
Krav til språk
Søknaden må skrives på engelsk. Protokollsynopsis og informasjon som dekkes av punkt 24, 59–73 i Annex I til Forordning (EU) Nr. 536/2014 (pasientinformasjon, samtykkeskjema, osv.) skal være skrevet på norsk.
Oversikt over språkkrav for de ulike medlemslandene i EU/EØS finnes i Questions and Answers Document - Regulation (EU) 536/2014 (PDF), Annex II.