Slik utvikles og godkjennes legemidler
Publisert:
|
Oppdatert:
Endringer
- : Lagt til lenker til prosedyrer for godkjenning av legemidler.
- : Endret lenke til intern nettside
- : Faglig gjennomgang. Oppdatert Norges satsingsområder i legemiddelsamarbeidet
- : Siden er omarbeidet. Utdypende tekst om det europeiske legemiddelsamarbeidet er flyttet inn på denne siden.
- : Inkludert informasjon om godkjenning av legemidler - endret tittel
Innhold på siden
Utvikling av legemidler
Nye legemidler kan komme fra grunnforskning, være basert på en innovativ idé, eller være resultat av forskning i industrielle miljøer. Utviklingsprogrammet strekker seg over flere år og må skje i samsvar med internasjonalt og nasjonalt regelverk.
Et viktig prinsipp ved gjennomføring av kliniske utprøvinger er etisk forsvarlighet og beskyttelse av forsøkspersonene. Dette har sin opprinnelse i Helsinkideklarasjonen(Ekstern lenke), og er stadfestet i retningslinjene for god klinisk forskningspraksis, ICH-GCP.
Klinisk utprøving av legemidler utføres for å skaffe til veie informasjon som til syvende og sist kan forbedre tilgangen til trygge og effektive legemidler til pasienter, samtidig som forsøkspersonene i studien er beskyttet.
- Les mer om klinisk utprøving av legemidler til mennesker
- Les mer om klinisk utprøving av legemidler til dyr
Faser i legemiddelutvikling
Legemidler til mennesker
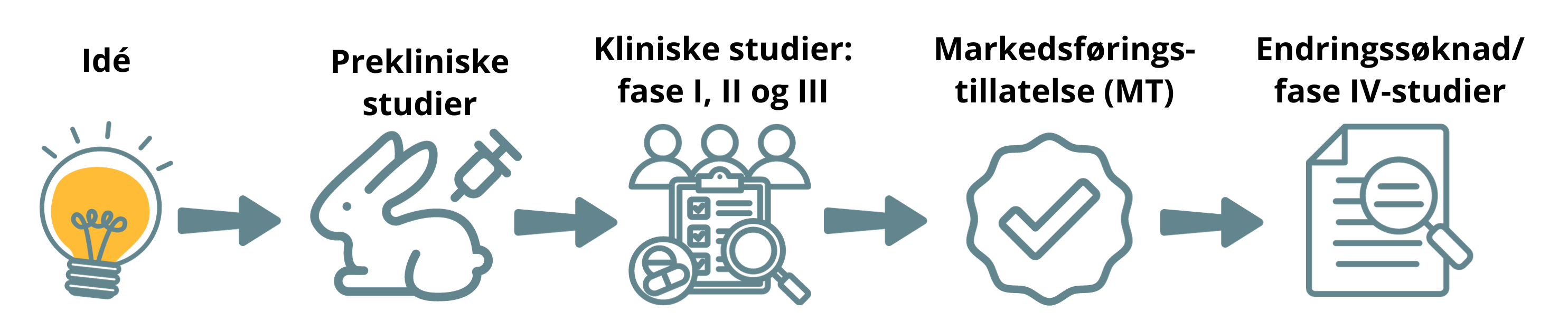
Prekliniske studier
Før legemiddelet prøves ut i mennesker, skal det gjøres mange forskjellige prekliniske studier. Dette er både in vitro-studier (studier utenfor en levende organisme – prøveglass) og in vivo-studier (dyrestudier). I dyrestudiene brukes det forskjellige dyrearter, og her undersøkes både preliminær effekt, toksisitet og farmakokinetikk.
Det utarbeides gjerne en klinisk utprøvningsplan for videre uttesting i mennesker når prekliniske studier viser at et nytt legemiddel ser ut til å kunne være effektivt og sikkert og det er bekreftet at det foreligger et medisinsk behov. Denne planen utarbeides sentralt i firmaet, eller i en forskningsgruppe. I de senere år er det blitt mer og mer vanlig å innhente vitenskapelige råd ("scientific advice") fra legemiddelmyndigheter.
Kliniske studier
Positive resultater (positivt risiko/nytte-forhold) fra et adekvat oppbygd klinisk studieprogram (klinisk fase I, II og III-studier) av høy vitenskapelig kvalitet er helt avgjørende for at et nytt legemiddel skal kunne få markedsføringstillatelse og bli gjort tilgjengelig for pasienter. I noen tilfeller vil det også være nødvendig å gjøre ytterligere kliniske studier (klinisk fase IV) etter at legemiddelet er kommet på markedet. De forskjellige fasene kan forstås på denne måten:
Fase 1: Humanfarmakologiske studier
Dette er første dose til mennesker, enten friske frivillige, eller til pasienter som har den aktuelle sykdommen. Behandlingen gis i et kort tidsrom og dose-toleranse, farmakokinetikk og farmakodynamikk undersøkes.
Fase 2: Terapeutisk eksplorative studier
Her undersøkes terapeutisk effekt hos et mindre antall pasienter. Det er korte dosefinnende studier med veldefinerte pasientgrupper. Det er ofte surrogatendepunkter i studien.
Fase 3: Terapeutisk bekreftende studier
Dette er større kontrollerte studier på effekt og sikkerhet i den aktuelle pasientgruppen. Studiene kan ha harde endepunkter og undersøker ofte dose-respons og, så langt det er mulig, bivirkningsmønster og frekvens. Det sammenlignes ofte med etablert behandling. Når fase 3 er avsluttet, har man som regel tilstrekkelig dokumentasjon til å kunne søke om markedsføringstillatelse (godkjenning) for legemiddelet. Fase 3-studier kan også være studier på allerede godkjente legemidler, men på en ny indikasjon.
Fase 4: Terapeutisk bruk
Slike studier kalles ofte "real life studier". Studiene gjelder legemidler som har markedsføringstillatelse, innenfor rammen av godkjent produktinformasjon (SmPC). Hensikten med disse studiene kan være å samle ytterligere informasjon om effekt, mortalitet, morbiditet, eller nye endepunkter. Hensikten kan også være å studere helseøkonomiske aspekter.
Legemidler til dyr
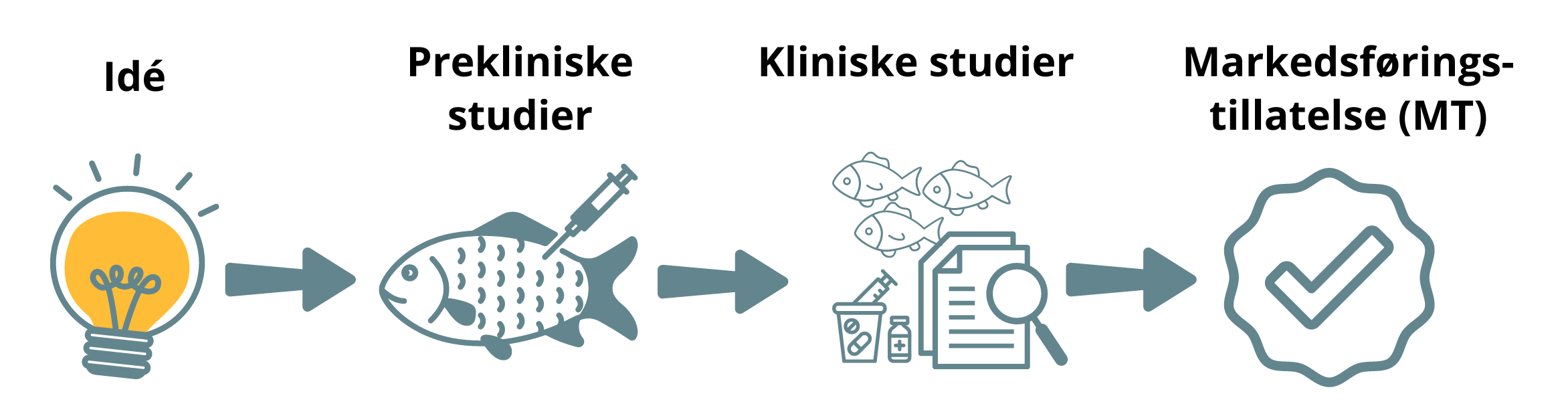
DMP tilbyr veiledning og råd til bedrifter og akademia når det gjelder legemiddelutvikling.
Godkjenning av legemidler
Godkjenning av et legemiddel innebærer at produsenten får markedsføringstillatelse (MT), det vil si tillatelse til å selge legemiddelet.
Et legemiddel blir bare godkjent for salg dersom legemiddelet har en nytte som overstiger risikoen ved bruk. Vurdering av nytte-/risikoforholdet til et legemiddel er basert på dokumentasjon som produsenten må sende inn når de søker om markedsføringstillatelse. I søknaden må produsenten dokumentere legemiddelets farmasøytiske kvalitet, sikkerhet og medisinske effekt.
Godkjenning skjer i all hovedsak gjennom det europeiske samarbeidet. Norge er del av dette gjennom EØS-avtalen.
Les mer om søknad og oppfølging av markedsføringstillatelse (MT)
Prosedyrer for godkjenning
Når produsenten skal sende inn en søknad om markedsføringstillatelse (MT) kan dette skje gjennom fire ulike prosedyrer. Det vanligste er at to medlemsland vurderer en søknad på vegne av hele EU/EØS. Deretter kan andre land kommentere vurderingen før legemiddelet eventuelt godkjennes for salg i alle medlemslandene. Dette kalles sentral prosedyre.
Les mer om de ulike prosedyrene:
Europeisk legemiddelsamarbeid
Godkjenning av legemidler i sentral prosedyre samordnes av vitenskapelige komiteer tilknyttet The European Medicines Agency (EMA) som holder til i Amsterdam. I dette legemiddelsamarbeidet stiller hvert land med utredningskompetanse, og godkjenningsarbeidet koordineres. Det vil si at kompetansen ikke ligger sentralt i EU-systemet. Vitenskapelige komiteer har en sentral rolle i EU-samarbeidet ettersom de vurderer utredningene som er gjort i hvert enkelt land og kommer fram til en felles beslutning, ofte ved avstemning. Ved beslutninger har hvert land en stemme, inkludert EØS-landene Norge og Island. EØS-landenes stemmer teller ikke, men gjengis i eget notat. På bakgrunn av komiteenes beslutning lages en innstilling som oversendes EU-kommisjonen som fatter endelig vedtak om å godkjenne et legemiddel. Dette kalles sentral prosedyre. Legemidler kan godkjennes i ulike prosedyrer.
Legemiddelsamarbeidet gjør at små land som Norge kan bidra med mye kunnskap og stille viktige spørsmål, og dermed påvirke beslutningen. Samtidig øker vår egen kompetanse på legemidler ved at vi drar nytte av andres kunnskap, som også er helt nødvendig for den nasjonale legemiddelforvaltningen. Store land har kapasitet til å behandle flere søknader. Et unntak fra dette mønsteret er Sverige, som prioriterer legemiddelspørsmål høyt og setter av ressurser til å utrede flere legemidler enn landets størrelse skulle tilsi.. Det er interessant at EU-samarbeidet gir små land mulighet til stor innflytelse dersom de satser på høy faglig kompetanse og kapasitet.
Norges satsingsområder i legemiddelsamarbeidet
Hvert år utreder Norge 10-15 % av nye søknader om markedsføringstillatelse i sentral prosedyre, og søknader om endret bruksområde for et legemiddel (indikasjonsendringer). Innenfor godkjenning av legemidler i legemiddelsamarbeidet har DMP valgt noen terapeutiske satsningsområder: Antibiotika, legemidler mot kreft, ATMP (Advanced Therapy Medicinal Product) og vaksiner til mennesker. For legemidler til dyr har vi et særskilt fokus på fiskevaksiner og antibiotika. Norge en forkjemper for en restriktiv linje når det gjelder antibiotikabruk, og har fått mye støtte for dette i EU. Årsaken er at den restriktive linjen har gitt oss langt mindre problemer med antibiotikaresistens enn vi ser i andre land vi kan sammenlikne oss med.
Forskjellige statuser for et legemiddel
I utgangspunktet skal alle legemidler som brukes i Norge være godkjent av DMP, slik at produsenten får markedsføringstillatelse (MT) for legemiddelet. Likevel kan legemidler med ulike statuser være i bruk – av ulike årsaker.
Les mer om ulike statuser for et legemiddel
Generisk og parallellimportert legemiddel
Det første godkjente legemiddelet med et nytt aktivt virkestoff kalles originallegemiddel. Når patenttiden går ut kan det utvikles kopier, såkalte generiske legemiddel.
Parallellimporterte legemidler er originallegemidler importert fra andre europeiske land der prisene er lavere enn i Norge.
Les mer om generisk legemiddel og parallellimportert legemiddel
Når blir et legemiddel tilgjengelig for bruk?
Når et legemiddel har fått markedsføringstillatelse (MT) og dermed er godkjent, kan legemiddelet markedsføres - settes på markedet. Dette er opp til selskapene om et legemiddel blir markedsført, og mange legemidler blir ikke markedsført i Norge.
Hvis et legemiddel skal betales av det offentlige, må myndighetene gjøre en kost/nytte-vurdering før det tas en beslutning om bruk.